Osteogenesis Imperfecta (OI)
Osteogenesis Imperfecta (OI) is a genetic disorder characterized, in most cases, by a defect in collagen formation, leading to increased bone fragility (high fracture risk) and skeletal deformities. The severity of OI varies widely: from mild forms with few fractures where the diagnosis may go unnoticed, moderate forms with multiple fractures, progressive bone deformities, and short stature, to very severe forms with in-utero fractures and death during the perinatal period. Other possible manifestations, though not in all patients, include dentinogenesis imperfecta, blue or gray sclerae, hearing loss, and joint hyperlaxity.
Approximately 80-90% of individuals with OI have mutations in one of the genes encoding the procollagen I chains: pro-α1 chain (COL1A1 gene) and pro-α2 chain (COL1A2 gene). In recent years, new genes associated with OI have been identified, which, depending on the gene, can present a dominant, recessive, or X-linked inheritance pattern.
Management of OI must be multidisciplinary and tailored to the degree of bone fragility and involvement.
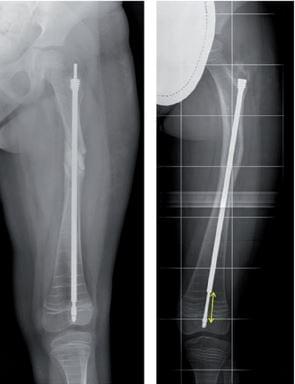
The orthopedic approach focuses on reducing the rate of pathological fractures and deformities. This includes the use of extendable intramedullary rods, which grow with the long bones of skeletally immature patients.
Cyclic therapy with bisphosphonates has become a key tool in treating moderate to severe OI, significantly improving bone mineral density and reducing fracture rates.